Embed Size (px)
Citation preview
E. Macromoléculas. Rev 27/11/2013
- 1 -
TEMA 8. PLEGAMIENTO DE PROTEINAS
Según la capacidad de las proteínas de adoptar plegamientos tridimensionales
definidos podemos clasificar a las proteínas en tres grupos:
Proteínas con una estructura tridimensional claramente definida.
Proteínas que pueden oscilar entre un número finitio de conformaciones
alternativas.
Proteínas intrínsicamente desectructuradas (Intrisically disordered
proteins). Se habla de IDP tanto en caso de proteínas que no tienen
ninguna estructura, como las que tienen un core estructurado pero gran
parte de la secuencia sin estructura.
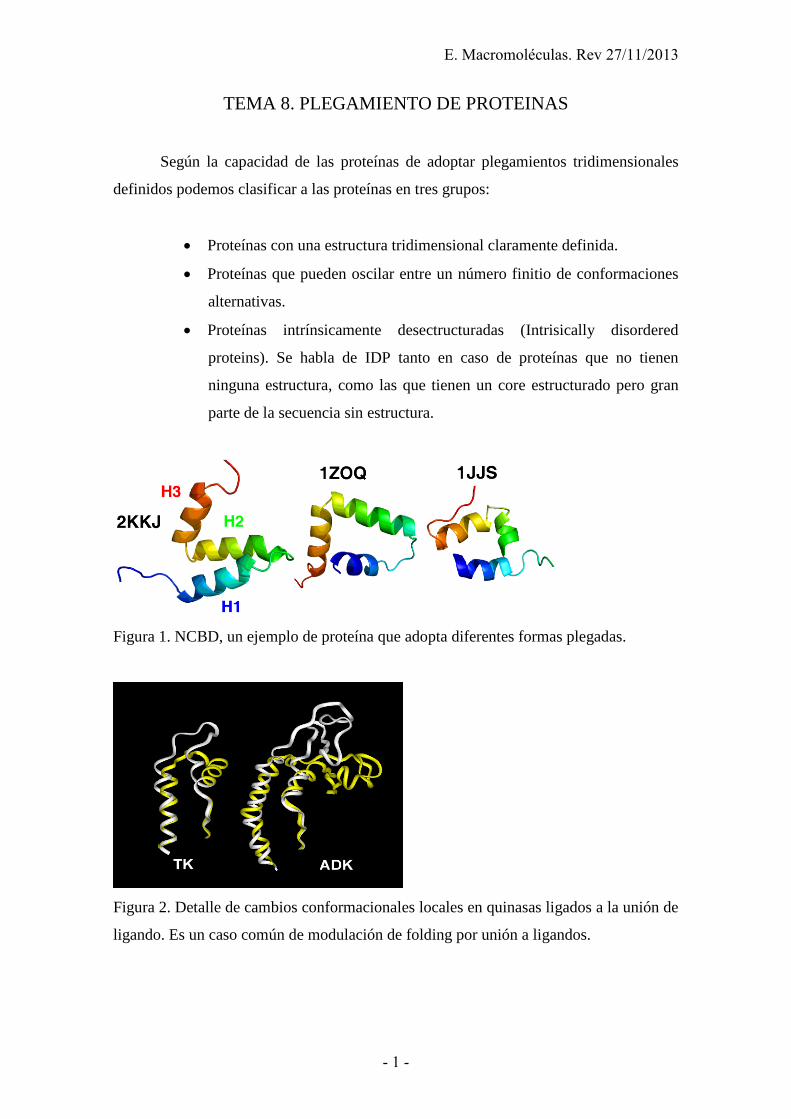
Figura 1. NCBD, un ejemplo de proteína que adopta diferentes formas plegadas.
Figura 2. Detalle de cambios conformacionales locales en quinasas ligados a la unión de
ligando. Es un caso común de modulación de folding por unión a ligandos.

E. Macromoléculas. Rev 27/11/2013
- 2 -
La estructura tridimensional de las proteínas (o su falta) está implícita en su
estructura primaria (su secuencia aminoácidica). Este hecho queda demostrado por el
gran número de proteínas que pueden desnaturalizarse y después renaturalizarse in vitro
sin la presencia de ningún cofactor o coayudante. Los recientes descubrimientos de la
existencia de numerosas proteínas que facilitan el plegamiento de las proteínas in vivo
(rotamasas, disulfide-isomerases, chaperones) no cambian nada el “dogma” central del
campo que es “que las proteínas contienen la información necesaria para replegarse”.
Las proteínas facilitadoras del plegamiento lo que hacen es simplemente catalizar, hacer
más rápido y eficiente el plegamiento, llevarlo hacia su final de manera más eficiente, al
i) mejorar la cinética de la ruta productiva del plegamiento y ii) evitar la formación de
estructuras no nativas.
Una interesante “excepción al dogma” lo representan las proteínas que pueden
adoptar dos o más estructuras. Estas proteínas “seleccionan” una de sus formas
accesibles en función del ligando (típicamente otra proteína) con la que interaccionan.
Con ello consiguen realizar varias funciones con una única secuencia (son lo que se
conocen como proteínas pluriemepleadas o “moolighting proteins”); si quereís ver una
discusión sobre estas proteínas ver por ejemplo (J.Am.Chem.Soc. (2011) 133, 12154-
12161)), o bien consiguen desencadenar respuestas complejas como consecuencia del
cambio conformacional ligado a la unión de un ligando.
Otra interesante “excepción al dogma” son las IDPs. Son proteínas, muy raras en
organismos inferiores, pero muy prevalente en organismos complejos que tienen la
totalidad o una parte importante de la secuencia sin estructura definida. Son proteínas
con secuencias muy repetitivas (de baja complejidad), pobres en aminoácidos apolares y
que precisan de otra macromolécula, típicamente otra proteína o un ácido nucleico para
plegarse. Pueden estar involucradas en “pluriempleo”, o en procesos complejos de
regulación (por ejemplo son comunes en proteínas de unión al DNA).
LA PARADOJA DE LEVINTHAL
El hecho de que muchas proteínas se plieguen solas de una manera tan
fidedigna, “independientemente” del medio celular y de un modo tan rápido es una

E. Macromoléculas. Rev 27/11/2013
- 3 -
paradoja aún no resuelta completamente, y formulada por primera vez por Levinthal.
Esta paradoja se formula de la siguiente manera: una cadena polipeptídica pequeña tiene
100 residuos, solo contando los ángulos y y contando que cada 30 grados se tiene
una conformación diferente tenemos para cada residuo 144 confórmeros y para la
proteína en su conjunto un total de 12200
confórmeros distintos. Si suponemos (y es una
suposición muy favorable) que cada 10 fs podemos cambiar de uno a otro confórmero
tendremos que para explorar todo el espacio conformacional la proteína necesitaría unos
10173
millones de años para plegarse. Un tiempo infinitamente superior a la edad del
universo. En realidad el tiempo de plegamiento de una proteína oscila entre el
microsegundo al minuto. Como toda paradoja ésta no tiene una solución obvia, y
durante muchos años ha existido un fuerte debate al respecto. A lo largo de este capítulo
veremos algunas ideas de cómo pensamos hoy que la proteína consigue resolverla.
La paradoja de Lenvinthal no pierde fuerza cuando pensamos en IDPs o en
proteínas con múltiples estructuras, ya que al final, sea sola, o en presencia de ligando la
proteína adopta una estructura tridimensional y lo hace de manera rápida y fidedigna.
Tampoco le resta fuerza la existéncia de maquinaria celular que acelera el plegamiento
de la proteína, porque la forma nativa continua siendo una propiedad intrínseca de la
proteína.
FUERZAS QUE CONDICIONAN EL PLEGAMIENTO DE LAS PROTEINAS
El plegamiento de una proteína debe conducir a un mínimo de energía libre, es
decir que la variación de energía libre a lo largo del plegamiento debe ser negativa:
Gfold) . Ignoramos si este mínimo es el más estable, o simplemente es uno muy estable
al que se llega de manera sencilla en el proceso de plegamiento.
La variación de energía libre en el proceso de plegamiento vendrá dada por el
balance de dos términos: el entálpico y el entrópico. El plegamiento debe por tanto
intentar optimizar la energía del sistema (i.e. debe mejorar las interacciones del sistema
proteína-solvente) y por otro lado debe hacer aumentar el desorden del universo.
Empezaremos describiendo las interacciones que marcan la estabilidad de las proteínas:

E. Macromoléculas. Rev 27/11/2013
- 4 -
Interacciones de enlace
La forma nativa de las proteínas presenta todas las distancias y casi todos los
ángulos próximos a los valores de equilibrio. La gran mayoría de las torsiones están
también en sus zonas estéricamente más favorecidas (e.j. mínimos mapa de
Ramachandran). Respecto a los enlaces amida en general se encuentran en la
conformación trans, con la excepción de las prolinas que a veces se encuentran en la
conformación cis. Es de destacar que el cambio de prolinas de conformación cis a trans
es muy importante in vivo y que incluso existen enzimas dedicado a catalizar este
proceso (las peptidyl-proline-isomerases o rotamases).
Unos enlaces covalentes que merecen especial atención son los puentes
disulfuro. Estos puentes se forman entre 2 cisteinas cuyas cadenas laterales se
encuentran próximas en el espacio. Los puentes disulfuros estabilizan en general la
estructura de la proteína como lo demuestra que las proteínas de vida extracelular
tengan una gran riqueza de los mismos. No obstante, en general los puentes disulfuro no
mejoran la cinética del plegamiento, ya que a lo largo del mismo se forman (y han de
romperse) puentes disulfuro no nativos y esto puede hacer muy lento el proceso de
plegamiento. De hecho, proteínas que se pliegan en microsegundos pueden tardar
minutos si existen posibilidades de formar puentes disulfuro no nativos en el
plegamiento. La naturaleza ha diseñado las Protein Disulfide Isomerases como un
mecanismo para facilitar la formación de los puentes disulfuro nativos y desestabilizar
los no nativos.
Interacciones no enlazantes
Son el conjunto de interacciones electrostáticas y de van der Waals que modulan
la interacción entre átomos no ligados por enlaces. Por tradición las interacciones no
enlazantes se clasifican en este campo en tres tipos en función de su teórica intensidad
en fase gas (medio anhidro).
Interacciones no específicas. Son un conjunto de interacciones de van der Waals
e interacciones electrostáticas débiles (en general interacciones de menos de 1 kcal/mol)

E. Macromoléculas. Rev 27/11/2013
- 5 -
que per se contribuyen poco a la estabilidad de la proteína, pero que en su conjunto son
muy importantes ya que son muy abundantes.
Los puentes de hidrógeno son interacciones básicamente electrostáticas de una
intensidad moderada (menos de 10 kcal/mol). Son muy direccionales y su existencia es
clave en todos los fenómenos de reconocimiento molecular y en los de formación de
estructura secundaria.
Las interacciones iónicas. También denominadas puentes salinos se forman entre
cadenas laterales de residuos cargados. Son totalmente interacciones electrostáticas,
tienen mucha importancia en el reconocimiento molecular a largas distancias y pueden
ser extraordinariamente intensas (superiores a 10 kcal/mol en fases apolares).
Durante muchos años se pensó que como en otros sistemas químicos, los
términos de enlace eran los que determinaban la estructura tridimensional de las
proteínas. Esto fue refutado por Alfinsen a principios de los sesenta (PNAS, 47, 1309
(1961)) cuando realizó un experimento clave que demostró que son las interacciones
no-enlazantes y no las enlazantes (puentes disulfuro) las que guían el plegamiento. Para
este experimento Anfinsen cogió ribonucleasa, una proteína pequeña (14000 Daltons) y
que tiene 4 puentes disulfuro. Desnaturalizó el enzima con urea y b-mercaptoetanol la
solución resultante la dividió en dos alícuotas P1 y P2. A la alícuotas P1 le eliminó
primero el -mercaptoetanol, dejo reposar y le eliminó luego la urea (alícuota resultante
P1bis). A la alícuotas P2 le eliminó primero la urea, y luego el -mercaptoetanol
(alícuota resultante P2b). Una vez hecho esto miro la actividad enzimática de P1bis y
P2bis. Encontró que la alícuota P2b había mucha actividad enzimática, mientras que en
P1b prácticamente no había nada. Esto demostraba que eran las interacciones no-
enlazantes y no las interacciones enlazantes las que guiaban el plegamiento de las
proteínas, pero no
determinaba cual de
ellas era la más
relevante.
urea
-Mercapto
-urea
-Mercapto
-Mercapto
-urea

E. Macromoléculas. Rev 27/11/2013
- 6 -
Figura 3. Esquema del experimento de Anfinsen. Rojo significa alícuota no activa, azul
activa
En un principio se pensó que eran los puentes salinos, dada su alta estabilidad
los responsables del plegamiento. Esta hipótesis está actualmente descartada por
diversos hechos: i) no todas las proteínas tienen puentes salinos, ii) el número de
puentes salinos que se forma en una proteína nativa es solo una porción de los que se
podrían formar, iii) los grupos cargados se encuentran mayoritariamente hacia el
exterior de la proteína, no hacia el interior que es donde prediciría la hipótesis de que
son claves. Además, cálculos teóricos han demostrado que la formación de puentes
salinos en agua está desfavorecida por el alto coste que supone desolvatar los iones que
forman el par. Recientemente, la mutagénesis dirigida ha demostrado que los puentes
salinos no son vitales para la estructura, ni aún en casos en los que están en el interior de
la proteína. En el experimento (Nature Structural Biology, 2, 122 (1995)) los autores
cogieron una proteína represor Arc que posee un puente salino interno doble (Arg31,
Glu36, Arg40) y mutaron los tres residuos por tres residuos apolares, en concreto por
Met, Tyr y Leu (respectivamente). La proteína resultante que ganaba contactos
hidropóbicos, pero que perdia el puente salino resultó ser mas de 4 kcal/mol más estable
que la proteína nativa!. En resumen, los puentes salinos pueden ser importantes para
mantener zonas concretas de proteínas, claves en el reconocimiento, y pueden ser
importantes en la guía del plegamiento, pero parece que no son los determinantes del
mismo.
Los puentes de hidrógeno son también candidatos razonables a tener un papel
clave en el plegamiento de las proteínas. Son abundantes, son claves en la estructura
secundaria, son relativamente intensos y muy direccionales. No obstante, parece
asumido que a pesar de su importancia, no son los puentes de hidrógeno los
determinantes del plegamiento, a pesar de su importancia. Esto es por dos razones
fundamentales: i) son interacciones demasiado direccionales y de corto alcance para
organizar el plegamiento global de la proteína, ii) si se cuentan los puentes de hidrógeno
totales de una proteína (proteína-proteína, y proteína-agua) se ve fácilmente que el

E. Macromoléculas. Rev 27/11/2013
- 7 -
número de puentes de hidrógeno totales no varia demasiado al producirse el
plegamiento de la proteína:
Así pues, no existe una fuerza única responsable del plegamiento de la proteína,
sino que dicho plegamiento depende de un conjunto de interacciones muchas de ellas de
escasa intensidad. Más importante aún para entender el plegamiento de una proteína es
pensar que la proteína es una cadena polipeptídica sumergida en agua, y que la
estructura de la misma cambia drásticamente a lo largo del proceso de plegamiento.
TERMODINAMICA DEL PLEGAMIENTO (ENTALPIA/ENTROPIA DE
PLEGAMIENTO)
El proceso de plegamiento de una proteína implica una variación del término
entálpico que se puede asociar a la variación de energía del sistema. La entalpía de
plegamiento dependerá pues del balance de la energía proteína-proteína, agua-proteína y
agua-agua. El término entrópico dependerá del grado de desorden que se consiga en el
proceso de plegamiento en la proteína y en el agua que la rodea.
Es sencillo ver que en el proceso de plegamiento se produce una mejora en las
interacciones proteína-proteína (p.ej se forman muchos puentes de hidrógeno entre los
residuos aminoacídicos) y que por el contrario el término entrópico es desfavorable ya
que la forma desnaturalizada “random-coil” está muy desordenada y la forma plegada
está muy ordenada. Si en principio tuviéramos 12200
estados (confórmeros) accesibles
en la forma desplegada, el costo entrópico de pasar a uno solo (forma plegada) sería
enorme (del orden de 200 kcal/mol). Esto no parece razonable, pero cual es entonces el
grado de desorden de la forma desplegada? Tradicionalmente se suponía que la forma
desplegada estaba muestreando una gran región del espacio configuracional accesible,
pero trabajos recientes de dinámica molecular y espectroscopía de NMR demuestran
que de hecho el estado desplegado es una combinación finita de un número limitado de
estados. La interconversión entre ellos es lo que da una imagen de desorden absoluto.
Esta nueva hipótesis sobre la entropía del estado desplegado tiene unas consecuencias
claras: al ser el número de estados accedidos por la forma desplegada más pequeño de

E. Macromoléculas. Rev 27/11/2013
- 8 -
lo que se esperaba, el coste entrópico del plegamiento de la cadena es también más
reducido, pero aun así es claro que la variación de entropía en la cadena es negativa y
que por lo tanto desfavorece mucho el proceso de plegamiento de la proteína.
Como se compensa el aumento de orden en la cadena polipeptídica?. Pues
gracias al aumento en el desorden el agua que rodea a la proteína. Esto nos lleva a
hablar del papel del agua en el plegamiento. El agua solvata tanto la forma plegada
como la desplegada de la proteína, por lo que es de esperar que juegue un papel
importante en el plegamiento. De hecho el agua modula el plegamiento de las proteínas
tanto a nivel entálpico, como a nivel entrópico. El agua forma puentes de hidrógeno con
ella misma y con todos los grupos polares de la proteína. En principio se formarán más
puentes de hidrógeno proteína-agua en la forma desnaturalizada, lo que podría inducir a
pensar que el agua desfavorece el plegamiento. No obstante, sabemos que el agua
favorece el plegamiento y de hecho remover agua con agentes hidrofóbicos es uno de
los mecanismos para desnaturalizar proteínas. Por que?
Cuando una cadena polipeptídica esta desordenada en agua sus grupos polares y
apolares se encuentran rodeados de moléculas de agua. El agua cuando está cercana a
los grupos apolares expuestos (lo que pasa en la forma desnaturalizada) no puede
interaccionar intensamente con ellas y reacciona formando estructuras poliédrica
altamente ordenadas alrededor del soluto. Estas estructuras tienen por objeto maximizar
la estabilidad intrínseca del agua entorno al soluto, pero tiene como inconveniente su
gran orden que hace que cuando estas estructuras se forman aumente el orden del agua.
Cuando 2 grupos hidrofóbicos se encuentran en solución cada uno de ellos forma uno
de estos poliedros de alta ordenación, con lo que la situación entrópica será peor. La
respuesta del sistema es la de comprimir los 2 grupos apolares (igual que 2 gotas de
aceite en agua) para que al juntarse el area accesible al solvente del conjunto se reduzca.
De esta manera los poliedros de alto orden se hacen más pequeños (que la suma de los 2
poliedros que existen antes de la fusión) liberándose en el proceso moléculas de agua
que aumentan el desorden del solvente. El efecto descrito se conoce como “efecto
hidrofóbico” y es el que hoy se asume que guía el plegamiento de la proteína hasta un
estado de colapso hidrofóbico (el molten globule), una forma compacta donde los

E. Macromoléculas. Rev 27/11/2013
- 9 -
residuos apolares ya están hacia el interior. La evolución de este estado compacto a la
forma nativa es entonces mucho más sencilla, ya que implica solo movimientos locales.
CINETICA DEL PLEGAMIENTO
La termodinámica del plegamiento es compleja de estudiar, pero aún lo es más
la cinética del mismo. La razón es clara: la escala temporal del proceso es mucho más
rápida que nuestra capacidad de medida. Así, se cree que muchas proteínas se pliegan
en escalas de tiempo de micro al milisegundo, y en muchos casos las etapas más
importantes pueden pasar en el lapsus del microsegundo, tiempos estos demasiado
pequeños para permitir el seguimiento experimental del plegamiento.
Así pues, el mecanismo cinético del plegamiento de una proteína dista de estar
claro. Durante mucho tiempo se asumió como cierto que existian solo 2 estados: el
nativo y el desnaturalizado. El paso de uno a otro sería vía un estado de transición y no
existirían intermedios de ningún tipo, al menos ninguno que un tiempo de vida
suficiente como para poder ser detectado experimentalmente. Como mucho se acepta,
dentro de este modelo la existencia de un estado privilegiado con un tiempo de
residencia significativo que sería el “molten globule”, que vendría a ser como una
estructura nativa desordenada, con los elementos fundamentales del empaquetamiento
ya definidos, pero carente de detalle fino en la estructura. La proteína se plegaría muy
rápidamente para llegar al “molten globule”, pero tardaría más en hacer las
reorganizaciones locales que darían lugar a la definición de la estructura terciaria nativa
de la proteína.
Figura 4. Perfíl energético típico de un modelo de plegamiento en dos estados.
E. Macromoléculas. Rev 27/11/2013
- 10 -
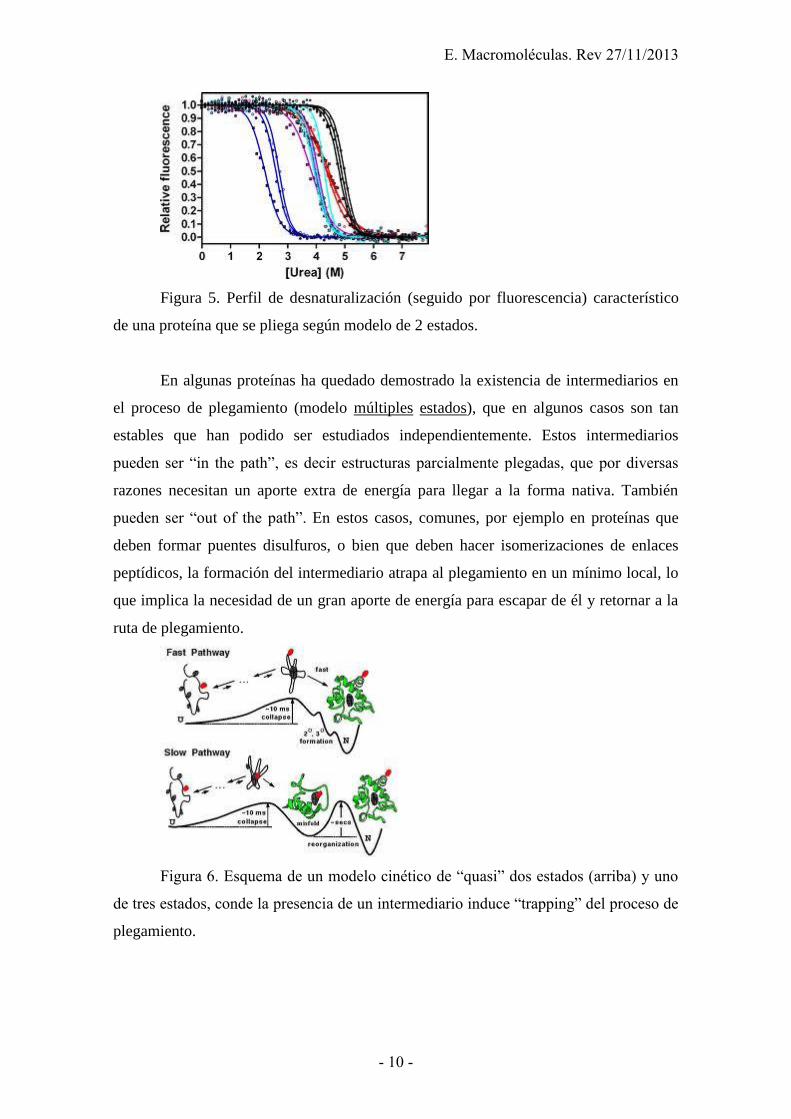
Figura 5. Perfil de desnaturalización (seguido por fluorescencia) característico
de una proteína que se pliega según modelo de 2 estados.
En algunas proteínas ha quedado demostrado la existencia de intermediarios en
el proceso de plegamiento (modelo múltiples estados), que en algunos casos son tan
estables que han podido ser estudiados independientemente. Estos intermediarios
pueden ser “in the path”, es decir estructuras parcialmente plegadas, que por diversas
razones necesitan un aporte extra de energía para llegar a la forma nativa. También
pueden ser “out of the path”. En estos casos, comunes, por ejemplo en proteínas que
deben formar puentes disulfuros, o bien que deben hacer isomerizaciones de enlaces
peptídicos, la formación del intermediario atrapa al plegamiento en un mínimo local, lo
que implica la necesidad de un gran aporte de energía para escapar de él y retornar a la
ruta de plegamiento.
Figura 6. Esquema de un modelo cinético de “quasi” dos estados (arriba) y uno
de tres estados, conde la presencia de un intermediario induce “trapping” del proceso de
plegamiento.

E. Macromoléculas. Rev 27/11/2013
- 11 -
Recientemente han ganado importancia las teorías del plegamiento en un estado
o “downhill folding”. Estas teorías dicen que la proteína existe únicamente en la forma
nativa y que las formas desplegadas no son más que excitaciones del mínimo. Es decir
que el plegamiento es un proceso expontáneo que pasa “colina abajo”, sin barrera de
activación hasta la forma nativa. La velocidad del proceso de plegamiento viene
entonces controlada simplemente por un término de difusión. Los apoyos de los
modelos de “downhill folding” vienen de estudios muy detallado de termogramas (por
ejemplo variación de coeficiente calorífico con la temperatura) de algunas proteínas.
Parece ser que es un mecanismo cinético correcto en un pequeño número de proteínas,
casi siempre muy ricas en alfa-helice, siempre pequeñas y siempre que se pliegan en
etapas de tiempo muy corto (entre el micro y el milisegundo).
Figura 7. Representación gráfica de un proceso de plegamiento según el paradigma de
“downhill folding”.
MODELOS DE PLEGAMIENTO
A lo largo del tiempo se han desarrollado modelos teóricos para visualizar como se
produce el plegamiento que intentan complementar lo que se puede describir por los
modelos cinéticos, que son en definitiva fenomenológicos.
Modelos clásicos de plegamiento:
I. Modelo del rompecabezas (jigsaw puzzle). Diferentes moléculas se pliegan de
manera diferente y solo convergen las rutas en el estado nativo.
II. Modelo del armazón (framework). Se forman los elementos claves de estructura
secundaria que difunden hasta dar la forma nativa

E. Macromoléculas. Rev 27/11/2013
- 12 -
III. Modelo clásico de nucleación. Una serie de residuos próximo nuclea, forma
estructura secundaria y alrededor de este nucleo se estructura la proteína.
IV. Modelo de colapso hidrofóbico. La proteína colapsa rápidamente por sus
residuos hidrofóbicos dando una estructura compacta que luego se
reorganizara para dar la forma nativa.
V. Modelo de nucleación-condensación. Se forma un nucleo difuso no estable, sin
estructura secundaria sobre el que colapsa la proteína formándose la
estructura secundaria y terciaria casi simultáneamente.
Recientemente y gracias en gran medida a simulaciones con modelos teóricos reducidos
se ha desarrollado un interesante modelo integrador denominado “entropy funnel”. De
acuerdo con este modelo:
• El plegamiento se entiende como un movimiento en un paisaje energético
(“energy landscape”).
• No hay una única ruta sino muchas que van convergiendo formándose diferentes
interacciones nativas
• Al ir avanzando se va reduciendo la amplitud del espacio conformacional de la
cadena (i.e. cada vez menos entropía) y se va mejorando la energía potencial.
• Es un modelo que permite integrar y generalizar características de diversos
modelos clásicos.
Conceptualmente podemos asimilar el plegamiento a un proceso en el que la proteína va
cayendo (de diversas maneras) en un embudo cada vez con menos libertad
conformacional, pero con más estabilidad energética.
Figura 8. Esquema simplificado del modelo de plegamiento en “entropy funnel”.

E. Macromoléculas. Rev 27/11/2013
- 13 -
El modelo del entropy funnel puede sofisticarse para englobar diferentes aspectos del
plegamiento y permite obtener una explicación razonable de la paradoja de Lenvinthal
ya que durante el plegamiento se va reduciendo el espacio configuracional accesible
disminuyendo en la práctica los estados accesibles en la práctica, lo que conduce a una
drástica reducción del tiempo teórico de plegamiento.
Figura 9. Generalización del modelo del entropy funnel para explicar plegamientos
complejos.





























